
Pancreatic Cancer Had No Druggable Target. Now It Does
Daraxonrasib in 2L PDAC: When the Data Actually Deliver

Pancreatic Cancer Had No Druggable Target. Now It Does.
Daraxonrasib in 2L PDAC: When the Data Actually Deliver
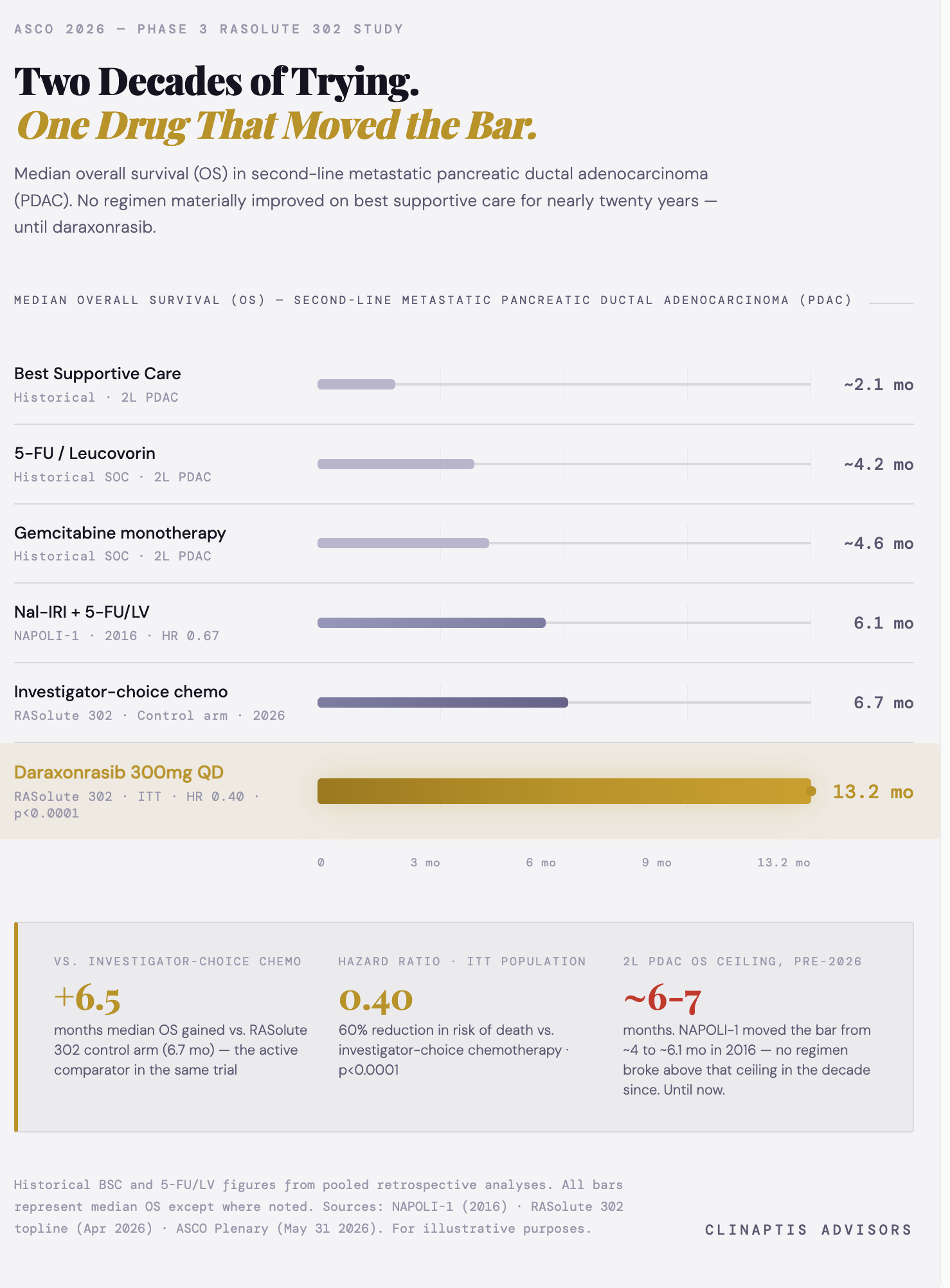
Pancreatic cancer has been one of oncology’s most stubborn problems — not for lack of trying, but because the biology kept winning. In the 2L PDAC setting, median OS (mOS) has hovered around 4-6 months for two decades, with every attempt at improvement running into the same wall: KRAS-driven tumors that shrug off chemotherapy, ignore immunotherapy, and have historically had no targetable driver to go after directly.
The KRAS story in PDAC has an underappreciated wrinkle. Much of the early clinical validation of RAS biology came from G12C inhibitors in lung cancer — but KRAS G12C represents only ~1-2% of pancreatic tumors. The alleles that actually define PDAC biology are G12D, G12V, and G12R — accounting for ~75-85% of all PDAC cases. A recent global tissue analysis of ~6,700 PDAC samples found ~90% of tumors carry RAS alterations addressable by a pan-RAS(ON) strategy, vs. just 1.4% for G12C-selective approaches.
Daraxonrasib was built for the right disease — targeting RAS in its active oncogenic state rather than downstream effectors. The result: HR 0.40 (p<0.0001), mOS 13.2 vs. 6.7 months vs. SOC chemotherapy, presented in the Plenary Session at ASCO 2026. At 12 months, 53% of daraxonrasib patients were alive versus 19% on chemotherapy. For those who’ve been following this story, this is the moment we were tracking.
Context: no 2L PDAC regimen had materially improved on ~6.1 months mOS (NAPOLI-1, 2016) in the decade prior.
The Data That Matter (Beyond the Headline)
HR 0.40 is the number that will dominate the coverage. It shouldn’t be the only number you look at.
The debate is no longer whether RAS inhibition works. The debate is what label that inhibition earns.
The wild-type question is binary and commercially consequential. RASolute 302 enrolled across RAS G12 variants and those without an identified RAS mutation — ~15-20% of enrolled patients. The Plenary data offers a nuanced read: WT patients were pooled with G13/Q61 mutations in a small subgroup (~41 patients total) — too underpowered for a definitive conclusion. But the more telling signal is this: G12 primary HR and ITT HR are essentially identical at 0.40. Non-G12 patients are not diluting the overall result — which suggests they’re deriving broadly similar benefit. The WT concern is reduced, not eliminated. An all-comers label remains plausible but unproven. In our view that distinction is still worth ~$2-3B in any credible peak sales range — and the label question won’t be definitively resolved until a powered WT-specific analysis exists.
The near-identical HR across G12 and ITT populations — 0.40 on OS in both — is the most important number in the full dataset. Benefit isn’t concentrated in G12 patients. It’s distributed.
The primary endpoint data is now disclosed — and it answers the question we were tracking. G12 primary OS HR: 0.40 (95% CI 0.30-0.54). G12 primary PFS HR: 0.45. ITT OS HR: 0.40. ITT PFS HR: 0.49. The G12 primary HR doesn’t exceed the ITT figure — it replicates it almost exactly. That’s the analytically important result: benefit isn’t concentrated in G12 patients, it’s broadly distributed across the enrolled population. The direction of the delta we flagged? Essentially zero — which is the most reassuring possible answer to the question. On ORR: daraxonrasib held at 33.2% in G12 patients versus ~35% in Ph1/2 — far less degradation than the ~9pp attenuation seen with sotorasib between Ph1/2 and Ph3. A quiet positive surprise in the full dataset. The control arm performed in line with historical benchmarks — mOS 6.6 months, mPFS 3.5 months, ORR ~11% — settling the pre-ASCO debate about whether investigator-choice chemotherapy artificially inflated the treatment effect. One design note: ~15% of chemotherapy patients did not initiate treatment, though all remained in the ITT analysis. At HR 0.40, this is difficult to view as a material driver of outcome.
The KM curve shape matters as much as the median. Post-ASCO commentary will anchor on mOS. The more important question is what the curves do over time. The NAPOLI-1 precedent was the cautionary case: early dramatic separation followed by attenuation — HR drifting from 0.65 to 0.73 between early and mature readout as the biology reasserted itself.
RASolute 302 tells a different story so far. Curves separate early — at 2-3 months — and show sustained divergence with little evidence of convergence through available follow-up. The 6-month PFS rate of 58.7% in G12 patients versus ~32% for chemotherapy reflects durable disease control, not just early tumor shrinkage. The curves look better than NAPOLI-1 suggested they would. But with median follow-up of only 8.5 months and fewer than one-third of daraxonrasib patients having experienced an OS event, the tail remains immature — and whether that divergence holds is the question follow-up data will answer.
Daraxonrasib hits the driver directly — a better mechanism than anything NAPOLI-1 had to work with. But PDAC is what George Sledge would call a biologically “smart” cancer — unlike CML, where imatinib was transformative against a single driver mutation, pancreatic cancer rarely relies on RAS alone. Co-mutations in TP53, SMAD4, and CDKN2A provide escape routes independent of RAS signaling. Broad RAS(ON) inhibition may suppress the dominant clone effectively — HR 0.40 suggests it does — but whether it delays or genuinely disrupts the underlying genomic complexity driving resistance is the open question the tail of the curve will eventually answer.
Regulatory and Commercial Path
An NDA submission is planned under the FDA Commissioner’s National Priority Voucher pathway. The agency has already signaled comfort: an Expanded Access Program is live following the FDA’s “safe to proceed” determination — patients are accessing daraxonrasib outside of a trial today, before formal approval.
Two days after the April 13 topline — stock having moved from ~$96 to ~$140 — RVMD priced a $2B concurrent offering: $1.5B equity at $142/share and $500M in 0.50% convertible notes due 2033, conversion price ~$199/share. Both tranches upsized. The convert strike sits ~107% above pre-data levels — a quiet institutional bet on where this stock goes over a 7-year horizon. Pipeline funded well beyond the 303 readout.
Consensus puts daraxonrasib PDAC-specific revenue at ~$7.5B by 2035 — less than half of projected company-wide sales of ~$15-16B. Nearly $8B of incremental consensus value sits in NSCLC, earlier-line PDAC, and additional RAS-driven tumors. The stock re-rated because 302 proved the mechanism at Ph3 scale — and consensus is extrapolating that proof across indications that haven’t read out yet. The more consequential post-ASCO question isn’t the 2L label. It’s whether that extrapolation holds as 301 and 303 mature.
RASolute 303 first dosed in April 2026 — three-arm global Ph3, ~900 patients, previously untreated metastatic PDAC regardless of RAS genotype: daraxonrasib monotherapy, daraxonrasib plus GnP, and GnP control. Combination arm uses a biweekly chemo schedule to preserve daraxonrasib dose intensity. Mature OS readout is a 2028 story at the earliest — the investment debate until then is fought on 302 data, the label question, and whatever interim signals 303 generates.
Second-line validates the mechanism. First-line determines the ceiling.
And the label question from 302 isn’t just regulatory housekeeping — an all-comers approval would structurally complicate the mutation-specific strategies every other RAS program is built around.
What We’re Watching
Three questions that matter more than anything being discussed on the post-ASCO call.
Toxicity at scale — better than feared, not fully resolved. The Ph3 data reframed the narrative: Grade ≥3 TRAEs of 43.6% with daraxonrasib versus 57.5% with chemotherapy, discontinuations of 1.2% versus 11.2%. Patient-reported outcomes favored daraxonrasib on both pain and QoL deterioration. The controlled trial picture is better than Ph1/2 suggested and better than SOC. The remaining question is community oncology practice — protocol-mandated monitoring, dose modification patterns, and real-world discontinuation rates in ECOG 2 patients who weren’t well-represented in the trial. The EAP cohort is where that answer begins to emerge. External discussant Jennifer Knox specifically highlighted pain and QoL outcomes as among the most clinically meaningful findings in the dataset — independent validation that the PRO data reflects genuine patient experience, not trial artifact.
Resistance kinetics. Curves show sustained divergence through 8.5 months median follow-up — better than the NAPOLI-1 precedent suggested. But PDAC’s co-mutations in TP53, SMAD4, and CDKN2A provide biological escape routes independent of RAS signaling. The timepoint at which KM curves begin to converge — if they do — will tell you whether daraxonrasib is buying durable remission for a meaningful subset or buying time for most. That question requires 18-24 months of follow-up data that doesn’t exist yet.
Franchise proof points: 303, 304, and beyond. The scientific risk around daraxonrasib has fallen materially. The valuation risk hasn’t — consensus embeds ~$15.8B total revenue by 2035 with roughly half from indications that haven’t read out yet. RASolute 304 (adjuvant PDAC) adds a third catalyst and potentially the largest addressable population of all three settings.”
RASolute 302 is a genuine landmark. The question that now replaces ‘does it work?’ is simpler and harder: how much of 303, 304, and the broader RAS franchise is already embedded in a $32B market cap?
Supplementary: The Downstream Commercial Consequence

This note is for informational purposes only and does not constitute investment advice. Clinaptis Advisors may hold positions in securities discussed.